 |
| Ilustración 2 Ácido oleico [2] |
 |
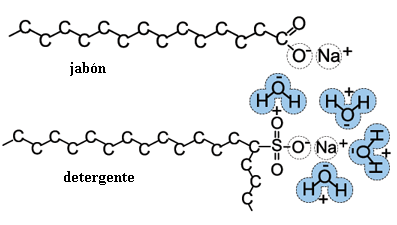
| Ilustración 2: Molécula de jabón [11] |
- Lípidos saponificables: estos se pueden separar en simples o complejos. Los simples se forman solamente por carbono, hidrógeno y oxígeno y los complejos so los que presentan además de los elementos mencionados en los simples fósforo, nitrógeno y azúfre. Dentro de estos se pueden mencionar los siguientes grupos:
Ácidos grasos: son ácidos orgánicos que se encuentran en las grasas. Tienen un número par de átomos de carbono, aunque se pueden encontrar excepciones a esto en los ácidos provenientes de la leche y grasa de animales rumiantes y se agrupan en forma de una cadena lineal. Es muy poco común encontrarlos en forma libre en los alimentos.
Se dividen en saturados, los cuales no presentan enlaces dobles entre los átomos de carbono; y los insaturados los cuales contrario a los saturados presentan enlaces dobles entre los átomos de carbono, estos en condiciones normales se presentan en estado líquido.
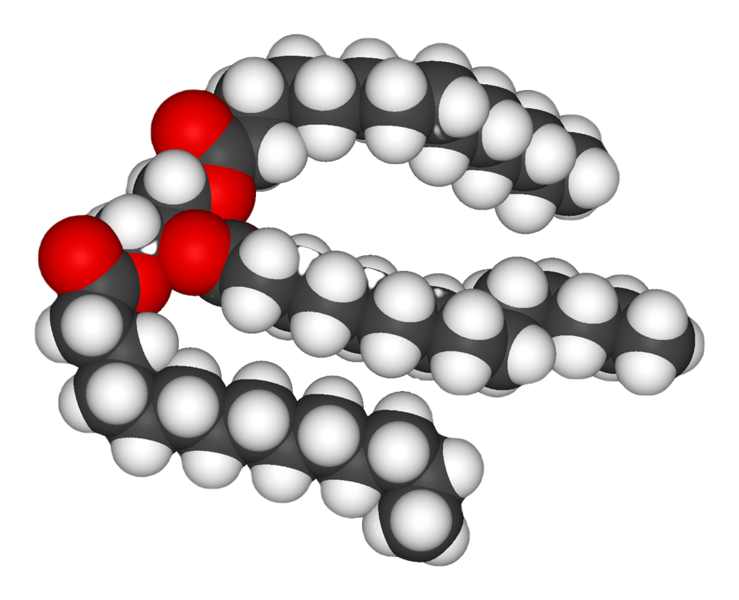
Acilglicéridos: son lípidos simples formados glicerol esterificado por uno, dos, o tres ácidos grasos [6], los cuales se denominan monoglicérdos, diacilglicéridos y triglicéridos respectivamente. En los seres vivos constituyen una gran reserva energética, en forma de grasa en los animales y de aceite en las plantas.
 |
| Ilustración 3: Representación de un triglicérido [7] |
Céridos: también llamadas ceras son sólidos a temperatura ambiente, pueden ser de origen animal o vegetal. Se forman por la esterificación de un ácido graso con un alcohol. Uno de los céridos más comunes es la cera de abeja. Presentan la característica de ser poco o nada solubles en agua.
Glucolípidos: son moléculas formadas por un lípido y un hidrato de carbono de cadena corta.
Fosfolípidos: estan compuestos por una molécula de glicerol la cual se une con dos ácidos grasos y un fosfato. Estos tienen dos partes, una que se asocia con el agua y otra que no. Algunas de las principales funciones de estos en el cuerpo son: activación de enzimas, formar parte de los ácidos biliares y ayudar a la síntesis de algunos compuestos.
- Lípidos insaponificables: entre estos se encuentran: eicosanoides, esteroides, y terpenos. [2]
Eicosanides: se originan mediante la oxidación de ácidos grasos, se componen de veinte átomos de carbono. Se dividen en tres grupos: prostaglandinas, tromboxanos y leucotrienos.
Terpenos: se forman debido a la polimerización del isopreno, su clasificación se basa en el número de isoprenos presentes en la molécula (dos, cuatro, ocho o más de ocho).
Esterioides: son derivados del hidrocarburo esterano. Dentro de este grupo se encuentran: vitamina D, hormonas sexuales y el colesterol.
Los lípidos cumplen varias funciones para los seres vivos, como lo son:
- Reserva de energía, la cual se almacena en los triglicéridos.
- Facilitan reacciones químicas.
- Regulación del metabolismo y reproducción mediante las hormonas, que son parte del los esteroides.
- Absorción de vitaminas como la A, D, E y K.
- Ayudan en el soporte estructural de las células.
Referencias
[1] Aula Virtual de Biología. Desconocida. Concepto y Clasificación. En linea. Fecha de consulta: 2/abril/2011. Disponible en: http://www.um.es/molecula/lipi00.htm
[2] Calleja García, J. 2010. Lipidos: características, funciones y clasificación. En linea. Fecha de consulta: 6/abril/2011. Disponible en: http://biologia.laguia2000.com/bioquimica/lipidos-caracteristicas-funcion-y-clasificacion
[6] Aula Virtual de Biología. Año desconocido. Acilgliceridos, grasas simples o neutras. En linea. Fecha de Consulta: 8/abril/2011. Disponible en: http://www.um.es/molecula/lipi02.htm
[7] http://upload.wikimedia.org/wikipedia/commons/thumb/6/64/Trimyristin-3D-vdW.png/739px-Trimyristin-3D-vdW.png Fecha de consulta: 6/abril/2011.




{kind=link}
{kind=link}